The mol3 file format
M. Louchart
q4md-forcefieldtools.org
Université de Picardie - Jules Verne, Amiens
P. Cieplak
q4md-forcefieldtools.org
Sanford Burnham Prebys Medical Discovery Institute.
F.-Y. Dupradeau
q4md-forcefieldtools.org
Université de Picardie - Jules Verne, Amiens
The Tripos mol2 file format presents many advantages (it handles force field atom types, atomic charges, atom and residue names, atom connectivitities, etc...). Consequently, this file format is the main output generated by the R.E.D. program and the R.E.D. Server server. The R.E.DD.B. database stores RESP and ESP charge values and force field libraries for small molecules and molecule fragments in this Tripos mol2 file format. The LEaP program available in the AmberTools and many programs (Jmol, VMD, Openbabel among many others) also recognize this file format.
However, in the concept of a force field library the Tripos mol2 file format has some limitations as well. Thus, when one wishes to build a polymer from a set of molecular fragments the Tripos mol2 file format does not handle information about the connecting atoms between molecular fragments (i. e. the head and the tail atoms of each fragment). Consequently, we have developed a new force field library file format, named "mol3 file format". This new file format is based on the Tripos mol2 file format to which information about the connecting atoms for molecular fragments can be added. In the future, this file format will be further developed, and it will be the main format used by R.E.D., R.E.D. Server and R.E.DD.B.
Using the mol3 file format as a force field library file format
-I.1- The LEaP program
-I.2- Using the Tripos2 mol2 file format in LEaP
-I.3- Using the mol3 file format in LEaP
-I.3.1- Features of the mol3 file format
-I.3.2- The "loadMol3" and "saveMol3" commands
-I.3.3- Demonstration of the use of the "loadMol3" and "saveMol3" commands
-I.4- Using the mol3 file format in other programs
-I.5- Download and install the patch requested for this tutorial in LEaP
-I.6- Polarizability values for the AMBER non-additive force field model

Using the mol3 file format as a force field library file format
In the context of a force field library two atoms belonging to a residue cannot share the same name, a residue is characterized by a single residue name and a single residue number, and the Cartesian coordinates available in a force field library do not really matter. Indeed, when a force field library is loaded in the LEaP program only the atom and residue names are used to match a residue in a PDB file, and this match is acheived independently of the residue conformation and/or configuration. Moreover, when some atoms are omitted in a residue in a PDB file LEaP can add the missing atoms based on these available in the force field libraries.
-I.1- The LEaP program
The LEaP program originally consisted of two programs, the xLEaP graphical interface and its text mode version tLEaP. A new non-graphical version of LEaP, named sLEaP, has been developed to try to solve some limitations and to enhance the features of xLEaP/tLEaP. Through its graphical interface, xLEaP remains particularly convenient for building and displaying structures and their related information (atom and residue names, force field atom types, charge values etc...). On the contrary, tLEaP is very efficient for automatically executing scripts in a text mode.
The new features presented in this tutorial are only related to xLEaP and tLEaP.
-I.2- Using the Tripos2 mol2 file format in LEaP
The Tripos mol2 file format is described here, and its use in the AmberTools is available in the corresponding manual. The q4md-forcefieldtools team has already implemented the "savemol2" command within the LEaP program in the past, and written a tutorial available here.
To demonstrate the advantages and limitations of the Tripos mol2 file format, we will build the ALA-ALA-ALA oligopeptide within xLEaP (a script can be written for the tLEaP version as well). To construct ALA-ALA-ALA, one needs three fragments for the alanine amino-acid, i. e. the N-terminal [(+)NH3CHCH3CO or "NALA"], the central (NHCHCH3CO or "ALA") and C-terminal [NHCHCH3COO(-) or "CALA"] fragments, and one needs to define the connecting atoms between these three fragments (i. e. the head and the tail atoms of each fragment). The three alanine fragments with their head and tail atoms as well as the ALA-ALA-ALA oligopeptide edited in the xLEaP program are presented in Figure 1.
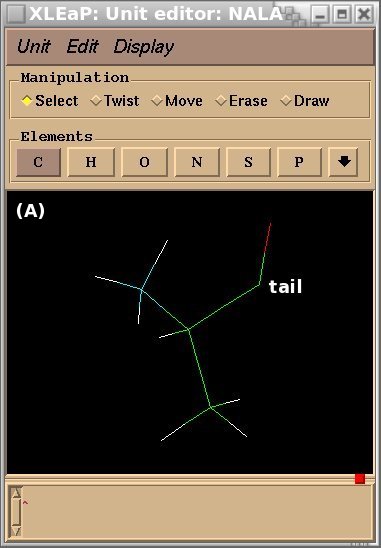
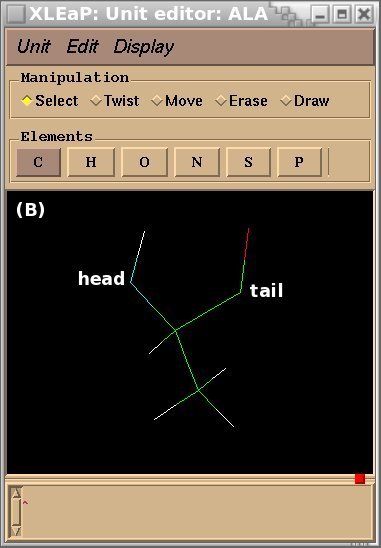
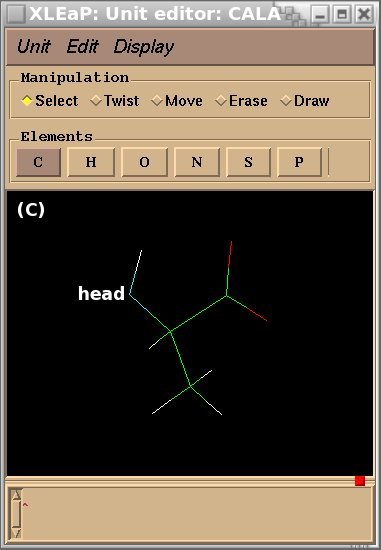
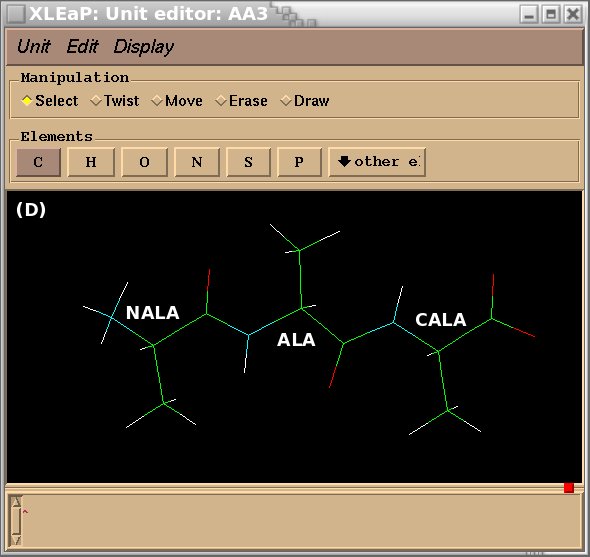
Figure 1
(A) residue NALA with its tail atom; (B) residue ALA residue with its head and tail atoms; (C) residue CALA with its head atom
(D) oligopeptide ALA-ALA-ALA built by using the NALA, ALA and CALA alanine fragments.
To build ALA-ALA-ALA the "sequence" command in the following LEaP script can be used:
# run xleap
xleap
# ...
# load the three force field libraries for the NALA, ALA and CALA fragments in the Tripos mol2 file format
NALA = loadmol2 NALA.mol2
ALA = loadmol2 ALA.mol2
CALA = loadmol2 CALA.mol2
# define the heads and the tails for NALA, ALA and CALA
# NALA: there is no head for NALA
set NALA tail NALA.1.C
set NALA.1 connect1 NALA.1.C
# ALA:
set ALA head ALA.1.N
set ALA tail ALA.1.C
set ALA.1 connect0 ALA.1.N
set ALA.1 connect1 ALA.1.C
# CALA: there is no tail for CALA
set CALA head CALA.1.N
set CALA.1 connect0 CALA.1.N
# create ALA-ALA-ALA by using the "sequence" command
AA3 = sequence { NALA ALA CALA }
# save the Tripos mol2 file for ALA-ALA-ALA
savemol2 AA3 AA3.mol2 1
This approach can be automatically executed by executing the following script:
tleap -f AA3-1.cmd
A limitation in this approach is that the head and tail atoms within fragments have to be manually defined as the corresponding information cannot be stored in the Tripos mol2 file format. This limitation can be overcomed when using the "OFF" Amber library file format. Indeed, the use of the OFF file format allows strongly simplifying the procedure as information about the connecting atoms between molecular fragments can be contained in the OFF file format. The corresponding script that uses this OFF file format is described below:
# run xleap
xleap
# ...
# load the three force field libraries for the NALA, ALA and CALA fragments in the Amber OFF file format
loadoff NALA.off
loadoff ALA.off
loadoff CALA.off
# create ALA-ALA-ALA by using the "sequence" command
AA3 = sequence { NALA ALA CALA }
saveoff AA3 AA3.off
This approach can be automatically executed by executing the following script:
tleap -f AA3-2.cmd
The OFF file format seems consequently attractive. However, a new problem encountered when using the OFF file format is that it is highly specific to Amber, and so far it cannot be visualized in graphical programs such as VMD or Jmol. On the contrary, the Tripos mol2 file format can be easily displayed in many graphical programs as described in Figure 2 below.
# load the mol2 file in VMD
vmd AA3.mol2
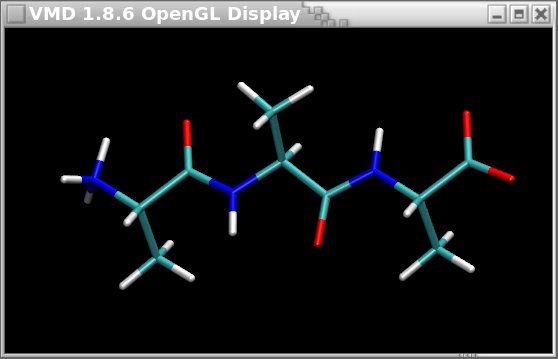
Figure 2
The ALA-ALA-ALA oligopeptide displayed in VMD
An ideal situation would be to have a force field library format that could be displayed in many graphical programs, and that could store information about connecting atoms between fragments.
-I.3- Using the mol3 file format in LEaP
-I.3.1- Features of the mol3 file format
We have combined some advantages of the Amber OFF and Tripos mol2 file formats by introducing the notion of connecting atoms between molecular fragments in the Tripos mol2 file format. The new file format obtained has been named the mol3 file format. Two new sections, named @<TRIPOS>HEADTAIL and @<TRIPOS>RESIDUECONNECT have been developed. They are simply added after the @<TRIPOS>SUBSTRUCTURE section in the Tripos mol2 file format, and they are presented in Table 1 below:
| @<TRIPOS>HEADTAIL | |
| A X | A: Atom name of the head; X: Residue number to whom the head belongs to |
| B Y | B: Atom name of the tail; Y: Residue number to whom the tail belongs to |
| @<TRIPOS>RESIDUECONNECT | |
| Z A B C D E F | Z: Residue number; A-F: Atom names of the connect0 up to connect5 atoms (line repeated for each residue) |
A-F and X-Z = 0 (zero) if null; by default the "head" is "connect0" and the "tail" is "connect1"
The following links provide examples of mol3 files for the NALA, ALA and CALA alanine fragments and the corresponding ALA-ALA-ALA oligopeptide.
We will continue to add new features in this force field library file format in a near future (see below section I.6).
-I.3.2- The "loadMol3" and "saveMol3" commands
The "loadMol3" command:
This command is used to load a mol3 file in LEaP. Once xLEaP or tLEaP is started, the syntax of the "loadMol3" command is the following:
variable = loadMol3 filename
The "saveMol3" command:
This command is used to save a molecule or a fragment in the mol3 file format. Once xLEaP or tLEaP is started, the syntax of the "saveMol3" command is the following:
saveMol3 unit mol3filename option
The "option" will affect the sixth column of the @<TRIPOS>ATOM section.
"0" will print chemical elements in the column, whereas
"1" will print force field dependant atom types; if the latters are not available, chemical elements will be printed instead.
This option has been introduced to generate Tripos mol2 file format fully compatible with R.E.DD.B.
Remark
The syntaxes of the loadMol3 and saveMol3 commands are identical to these of the loadMol2 and saveMol2 commands, respectively.
The syntax of each command available in xLEaP (or tLEaP) can be displayed by typing the "help" command at the prompt message. To get help about "loadmol3" and "savemol3", use the following commands:
help loadMol3
help saveMol3
-I.3.3- Demonstration of the use of the "loadMol3" and "saveMol3" commands
Mol3 files could be used as it follows:
# run xleap
xleap
# ...
# load the three force field libraries for the NALA, ALA and CALA fragments in the mol3 file format
# Any type of file name and file extension can be used for Mol3 files
# (the names/extension 'XXX'-mol3.mol2 reported here are used for pedagogical purposes only)
NALA = loadmol3 NALA-mol3.mol2
ALA = loadmol3 ALA-mol3.mol2
CALA = loadmol3 CALA-mol3.mol2
# create ALA-ALA-ALA by using the "sequence" command
AA3 = sequence { NALA ALA CALA }
savemol3 AA3 AA3-mol3.mol2 1
Once loaded in LEaP, mol3 files can be easily converted into OFF, Tripos mol2 and PDB files:
saveoff AA3 AA3.off
savemol2 AA3 AA3.mol2 1
savemol2 ALA ALA.mol2 1
savemol2 NALA NALA.mol2 1
savemol2 CALA CALA.mol2 1
savepdb AA3 AA3.pdb
-I.4- Using the mol3 file format in other programs
Considering that the Tripos mol2 file format is highly similar to the mol3 file format, the later should be recognized by other programs. For instance, the vmd program can display mol3 files when the ".mol2" extension is used (see Figure 3).
# load the mol3 file in VMD
vmd AA3-mol3.mol2
Figure 3
The ALA-ALA-ALA oligopeptide displayed in VMD
Mol3 files can also be displayed in Jmol (see the Java applet available here).
-I.5- Download and install the patch requested for this tutorial in LEaP
Before downloading the LEaP files requested for this tutorial, each user should check the LEaP license in the AmberTools. Each user of the LEaP source code files downloaded from q4md-forcefieldtools.org have to strictly follow the license defined on the Amber website.
To be used the patch downloadable here as well as the mol3File.c and mol3File.h files have to be applied to the AmberTools 1.5 by executing:
cp mol3File.c mol3File.h $AMBERHOME/AmberTools/src/leap/src/leap/
cp LEaP_q4md-forcefieldtools-Mol3.patch $AMBERHOME
cd $AMBERHOME
patch -p0 -N < LEaP_q4md-forcefieldtools-Mol3.patch
This feature is included within the AmberTools versions 12 - 16 (no need to apply this patch).
-I.6- Polarizability values for the AMBER non-additive force field model
Recent versions of the PyRED program (or R.E.D. Python) available at R.E.D. Server Development generate atomic polarizability values in the mol3 file format: atomic polarizabilities are required in the Amber polarizable force field (non-additive force field model). A column of polarizability values is printed after the column of atomic charges in the @<TRIPOS>ATOM section. See below examples of mol2 (without polarizability value) and mol3 (with polarizability values) files generated by PyRED:
Alanine central fragment in the mol2 file format (without polarizabilities; for the AMBER additive force field model):
ALA.mol2
Alanine central fragment in the mol3 file format (with polarizabilities set to zero; for the AMBER additive force field model):
ALA-mol3.mol2
Alanine central fragment in the mol3 file format (with polarizabilities set to values required for the AMBER non-additive force field model):
ALA-mol3.mol2
It is important to underline, that the actual LEaP version does not handle polarizabilities in the mol3 file format yet (to be implemented).
If you have questions about this tutorial, please, send your emails to the q4md-forcefieldtools mailing list. We will answer to the queries about the q4md-forcefield tools in the Amber or CCL mailing lists as well.
Last update of this tutorial page: January 12th, 2012.
Charge derivation data free for download.
Université de Picardie Jules Verne. Sanford Burnham Prebys Medical Discovery Institute.
© 2009-2024. All rights reserved.
