Introduction
Cyclodextrins (CDs) are macrocyclic oligosaccharides composed of
D-glucose units linked together by α(1→4) glycosidic bonds. CDs are versatile compounds with various applications in chemistry and biology: they can be used as building blocks for supramolecular structures and/or act as drug/ligand carriers as one of their ability is to stabilize soluble organic molecules in liquid phases. To affect their solubility and/or their selectivity towards specific targets, CDs are substituted. Derivatization of native CDs can be of several types: (i) substitution with common organic protecting groups to modify the solubility properties and (ii) grafting of ligands such as amino-acid derived arms to modulate recognition mechanisms. In the absence within the GLYCAM force field of the fragments required to build the aforementioned substituted CDs, a new Force Field Topology DataBase (FFTopDB) compatible with the reported CD based molecular systems has been developed. In this work, the considered substitutents are heterogeneous,
i. e. of organic, peptidic and carbohydrate nature.
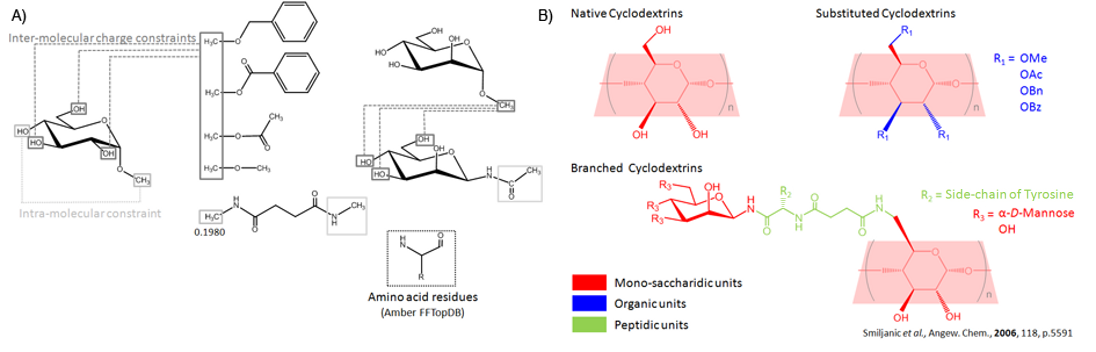 Scheme 1
Scheme 1
Computational details
Multiple molecules, multiple conformations and multiple molecular orientations were used during the charge calculation procedure. RESP charge derivation and force field library building for the presented fragments were carried out in a single R.E.D. IV job following the procedure summarized on Scheme 1 and corresponding to a 34-structure RESP fit. Eight molecules, namely methyl α-
D-glucopyranoside, β-
N-acetamido-
D-mannoside, methyl α-
D-mannoside, dimethyl ether, methyl acetate, benzyl methyl ether, methyl benzoate and
N,N'-dimethylsuccinamide were considered. Two conformations were selected and optimized for the sugar-derived units since Glucose and Mannose monosaccharide derivatives show two main populations in solution relative to a
gg/gt isomerism around the the ω dihedral angle. Four molecular orientations based on the (C1 C3 C5) and (C2 C4 O5) sets of atoms were considered in molecular electrostatic potential (MEP) computation.
[1] For each of the protecting groups/ligands, only one conformation corresponding to the lowest minimum obtained after geometry optimization was taken into account. Two orientations, based on the connected C(Me)-C-O atoms for the organic protecting groups and based on the (C1 C2 C3) atoms for
N,N'-dimethylsuccinamide were considered.
[1] No weighting factor to bias a conformation over another was applied during the charge derivation procedure. Inter- and intra-molecular charge constraints were used during the fitting step to define the required molecular fragments. Inter-molecular charge constraints were set to a target value of zero between (i) the 2-hydroxyl, 3-hydroxyl and 6-hydroxyl groups of the methyl α-
D-glucopyranoside and the methoxy group belonging to the different organic protecting groups and (ii) the 3-hydroxyl, 4-hydroxyl and 6-hydroxyl groups of the β-
N-acetamido-
D-mannoside and the methyl group of methyl α-
D-mannoside. Intra-molecular charge constraints between the 4-hydroxyl and the methyl group of the methyl α-
D-glucopyranoside as well as for one of the
NH-methyl group of
N,N'-dimethylsuccinamide and the acetyl group connected to β-
N-acetamido-
D-mannoside were set to zero. For the sake of compatibility between organic/sugar and peptidic/sugar connections, an additional intra-molecular charge constraint set to a value of 0.1980 was imposed for the methyl group of
N,N'-dimethylsuccinamide (that not already involved in the intra-molecular charge constraint previously defined). Geometry optimization and MEP computation were carried out with the Gaussian03 program, while charge fitting was done using the RESP program. Geometry optimization was carried out at the RHF/6-31G** level of theory, whereas MEP computation was performed at the RHF/6-31G* one using the Connolly surface algorithm. The molecular orientation of optimized geometries was controlled before MEP calculation using the rigid-body reorientation algorithm implemented in R.E.D. leading to highly reproducible charge values. RESP charge fitting was carried out following a two-stage fitting procedure with a hyperbolic restraint function, using a weighting factor of 0.0005 and 0.001 for the two stages, respectively. A RRMS (Relative Root Mean Square) value of 0.096 was obtained for the charge fitting step between the MEP calculated by quantum chemistry and that generated using the derived charge values.
Discussion
CDs substituted with organic protecting groups or with an amino-acid derived arm can be considered like heterogeneous systems as they display carbohydrate, organic and peptidic components. The modeling of such a molecular system should be heterogeneous as well and different force fields specific to each of these constituents are commonly used. The GLYCAM approach for deriving atomic charges has proven to be highly innovatrice and effective: the weighting of the different conformations taken into account during the charge fitting step is related to their occurence during molecular dynamics (MD) simulations.
[2] However, this approach applied to study the conformational space of a ligand to be grafted on a CD remains a complex task. Consequently, considering that the highly heterogeneous CDs studied in this work are of a “glyco-organo-peptidic” nature, we have chosen to derive RESP charges following the “Amber” approach as a single and homogeneous procedure. The charge derivation and force field library building procedures only involve representative minima optimized by quantum mechanics in the gas phase followed by a MEP computation step using the Connolly surface algorithm and a two RESP-charge fitting stage.
[3] Furthermore, for the sake of consistency and homogeneity, we developed a unique force field, namely “q4md-CD”, to model these CD based systems. q4md-CD presents the following features: (i) atom charges for all required fragments were derived following the procedure previously reported. The glycocluster FFTopDB is available as a suite of force field libraries for molecular fragments in the Tripos mol2 file format and is used to build Amber OFF force field libraries. A LEaP
script is available for this purpose in this project. (ii) A minimal number of charge constraints was used during the fitting step to keep the bias on the RRMS as low as possible, and consequently charge values of aliphatic hydrogen atoms were not constrained to the zero value. (iii) Scaling factor values of 1.2 and 2.0 for the 1-4 electrostatic and van der Waals interactions were used in MD simulations, respectively. (iv) Geometrical parameters are adapted to the nature of the considered ligand. Force field parameters used in association with the FFTopDB reported are described in the
frcmod file. (v) Compatibility with amino acid residues from the Amber FFTopDB is consequently assured.
Conclusion
Derived charge values have been validated after analyses of 50 nsec MD simulations. Structural characteristics (hydrogen bond patterns, ω dihedral populations, sugar pucker and CD solvation) observed during MD compare well to experimental data. To the best of our knowledge q4md-cd represents the first development of an Amber-GLYCAM hybrid force field, which allows studying glycoconjugates in a fully homogeneous approach. Extension of the force field to more complex glycoconjugates is underway. The corresponding study will be included in a forthcoming paper.
[1] Molecular orientations for monosaccharide units are based on the (C1 C3 C5), (C5 C3 C1), (C2 C4 O5) and (O5 C4 C2) sets of three atoms. Molecular orientations for the organic protecting groups are based on the: (i) (CX OS CM) and (CM OS CX) sets of three atoms for dimethyl ether, (ii) (CX OS C) and (C OS CX) sets of three atoms for methyl acetate, (iii) (CX OS CM) and (CM OS CX) sets of three atoms for benzyl methyl ether, and (iv) (CX OS C) and (C OS CX) sets of three atoms for methyl benzoate. Molecular orientations for N,N'-dimethylsuccinamide are based on the (C1 C2 C3) and (C3 C2 C1) sets of three atoms. See the corresponding PDB files for atom naming convention.
[2] Woods, R. J.; Chappelle, R. J. Mol. Struct. (Theochem) 2000, 527, 149-156. Basma, M.; Sundara, S.; Calgan, D.; Varnali, T.; Woods R. J. J. Comput. Chem. 2001, 22, 1125-1137. Kirschner, K. N.; Yongye, A. B.; Tschampel, S. M.; Gonzalez-Outeirino, J.; Daniels, C. R.; Lachele Foley, B.; Woods, R. J. J. Comput. Chem. 2007, 29, 622-655.
[3] Bayly, C. I.; Cieplak, P.; Cornell, W. D.; Kollman, P. A. J. Phys. Chem. 1993, 97, 10269-10280. Cornell, W. D.; Cieplak, P.; Bayly, C. I.; Kollman, P. A. J. Am. Chem. Soc. 1993, 115, 9620-9631. Cieplak, P.; Cornell, W. D.; Bayly, C. I.; Kollman, P. A. J. Comput. Chem. 1995, 16, 1357-1377.






