The β-hematin crystal consists of head-to-tail hematin dimers or cyclic dimers of ferriprotoporphyrin IX coordinated
via Fe-O propionate bonds (FPD).
[1] In order to be able to study by molecular dynamics (MD) simulations the structure and the dynamics of the various stereoisomers of FPD reported in the literature as well as analogs of interest and oligomeric clusters observed during the early stage of crystallization a new Amber type additive force field was generated for the molecules of interest. Atomic charge derivation, atom typing, force field library building and force field parameter generation were carried out by using the PyRED program available at the R.E.D. Server Development Internet site.
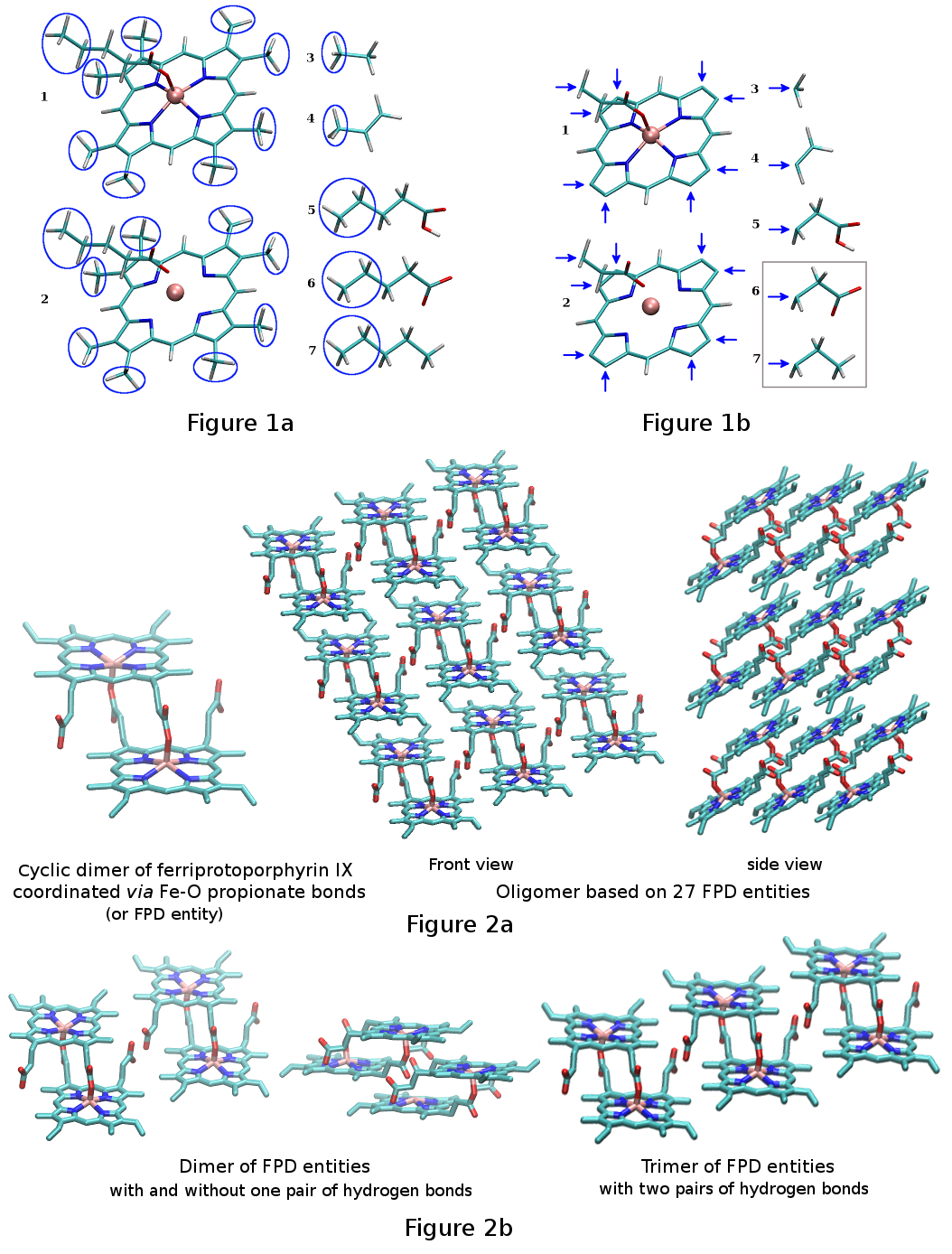
[2] This empirical work was achieved on the basis of six different input molecules or elementary building blocks (Figure 1a): a well-designed monomeric derivative of heme and a list of five organic molecules: ethane, propene, pentanoic acid, pentanoate and pentane. The porphyrin ring of the monomeric heme derivative presents methyl capping groups in place of the characteristic vinyl and propionic acid groups of heme B, and contains a ferric iron, which is coordinated by the four heminic nitrogen atoms and a fifth pentanoate ligand. This heme derivative was repeated twice in the list of input molecules allowing the consideration of both a covalently bound ferric ion bearing a non-integer charge value, and an ionically bound ferric ion with an integer charge value of +3 e. FPD stereoisomers and analogs were constructed from empirical molecular fragments, which were issued from the monomeric heminic molecule, that was duplicated and from the organic molecules: after removing a methyl or an ethyl capping group ethane, propene, pentanoic acid, pentanoate and pentane were used as donors of methyl, vinyl, 2-carboxyethyl, 2-carboxylatoethyl, and propyl groups for the porphyrin ring of the heme derivative, respectively. The porphyrin ring was used as an acceptor of these molecular fragments removing its eight capping methyl groups and the ethyl group of the pentanoate ligand (Figure 1b).
Quantum mechanical computations were achieved by using the Gaussian 2009 program (version D.01).
[3] For the heme building block the high spin state (S = 5/2) of Fe(III) was considered in density functional theory calculations.
[4] The geometries of the different input molecules considered in this work were optimized by using the 6-31G* basis set and the B3LYP method.
[5] For each building block the lowest minimum found after a conformational search was selected. To account for the dimeric structure of the FPD entity the geometry of the monomeric heme building block was optimized with constraints: the positions of the α-carbon atom of the pentanoate ligand and of the two closest heminic nitrogen atoms to this α-carbon atom were frozen during geometry optimization.
[6] These constraints were chosen because they do not lead to any imaginary frequency, and yielded an optimized structure, which was close to that observed in the di-heminic crystal structure. The wave function of each minimum was checked: if found unstable it was optimized and the geometry of the corresponding minimum was optimized again using the newly determined wave function. Molecular electrostatic potentials (MEP) were also computed at the B3LYP/6-31G* theory level, and involved the Connolly surface algorithm with the default density of points and a radius of 1.8 Å for the iron atom. For each optimized geometry a pair of molecular orientations was automatically selected based on a rigid-body reorientation algorithm ensuring the control of the orientation of the geometry involved in MEP computation, and consequently the reproducibility of the atomic charge values.
[7] To further check each optimized geometry frequencies were also computed at the B3LYP/6-31G* theory level.
RESP atomic charge fitting was carried out using a standalone version of the RESP program, and following the standard two-stage procedure.
[8] The molecular fragments required for the construction of the FPD stereoisomers and analogs were designed by setting specific intra-molecular charge constraints during the charge fitting step for the capping groups. For each elementary building block a single intra-molecular charge constraint with a value of zero was applied to the alkyl capping groups. More particularly the atomic charges of the eight methyl capping groups of the porphyrin ring and of the ethyl capping group of the ethanoate ligand were constrained to have a total charge of zero.
[9] The atoms involved in these intra-molecular charge constraints were removed from the molecule to yield the required molecular fragments and the corresponding force field libraries to the Mol3 file format.
[10] This approach was found to be the one, that lead to the lowest relative root mean square (RRMS) value and to the highest correlation coefficient (r2) value for the charge fitting step. An additional intra-molecular charge constraint of +3.0 e was used to define the ionically bound iron atom for a heme derivative. RRMS and r2 values of 0.057 and 0.996 between the MEP calculated by quantum chemistry and that generated using the derived charge values were obtained for the charge fitting step, respectively. Highly similar values were obtained in the absence of intra-molecular charge constraints. The small RRMS value and high r2 value obtained for this two stage charge fitting step as well as the small differences of RRMS and r2 values between the charge fitting steps carried out with and without intra-molecular charge constraints demonstrate the accuracy of the fitting step performed in this study, and the weak effect of these constraints.
[8]
Atom types and force field parameters for the input molecules were determined by the PyRED program based on an internal dictionary of atom types and a database of force field parameters corresponding to the Amber99 force field.
[11] Missing force field parameters for the organic parts of the input molecules were determined by analogy to existing ones. Force field parameters involving the covalently bound ferric ion were determined as it follows: (i) bond and angle equilibrium values were taken from the crystal structure (
Cambridge Structural Database code: AYILIE),
[12] (ii) bond and angle force field constants were calculated from the Hessian matrix (using the Gaussian formchk file obtained from frequency computation) and by applying the Seminario's method,
[13] (iii) the energy barrier of the dihedral force field parameters were set to zero, (iv) improper force field parameters were not considered, and (v) van der Waals parameters for the Fe(III) atom were taken from the Amber 12 program contribution (R* = 1.2 Å, ε = 0.05 kcal/mol).
[14] Force field parameters were combined into frcmod files.
[15] The additive q4md-forcefieldtools force field generated in this work, and reported in this R.E.DD.B. project was exported within the LEaP program using a dedicated script to generate parameter/topology and Cartesian coordinate files.
[16] This new force field was validated by using MD simulations in condensed phase and the pmemd program:
[14] the structures and the dynamics of the FPD entity as well as a polymer of twenty-seven FPD entities were studied in condensed phase (Figure 2a).
[17] Results were found to be in good agreement with experimental data and with the crystal structure of β-hematine. This force field was finally used to study small clusters of the β-hematin crystal (Figure 2b).
[2] F.-Y. Dupradeau, A. Pigache, T. Zaffran, C. Savineau, R. Lelong, N. Grivel, D. Lelong, W. Rosanski, P. Cieplak, Phys. Chem. Chem. Phys. 2010, 12, 7821; E. Vanquelef, S. Simon, G. Marquant, E. Garcia, G. Klimerak, J. C. Delepine, P. Cieplak, F.-Y. Dupradeau, Nucl. Acids Res. 2011, 39, W511; F. Wang, J.-P. Becker, P. Cieplak, F.-Y. Dupradeau, PyRED: Object oriented programming for Amber force fields, Université de Picardie - Jules Verne; Sanford | Burnham Medical Research Institute, Nov. 2013.
[3] Gaussian 09, Revision D.01, M. J. Frisch et al. Gaussian, Inc., Wallingford CT, 2009, and http://www.gaussian.com.
[4] D.S. Bohle, P. Debrunner, P.A. Jordan, S.K. Madsen, C.E. Schulz, J. Am. Chem. Soc. 1998, 120, 8255. The spin multiplicity of each input molecule is reported in the PyRED 'Project.config' configuration file, which available for download here.
[5] V.A. Rassolov, J.A. Pople, M.A. Ratner, T.L. Windus, J. Chem. Phys. 1998, 109, 1223; V.A. Rassolov, J.A. Pople, M.A. Ratner, P.C. Redfern, L.A. Curtiss, J. Comp Chem. 2001, 22, 976; K. Kim, K.D. Jordan, J. Phys. Chem. 1994, 98, 10089; P.J. Stephens, F.J. Devlin, C.F. Chabalowski, M.J. Frisch, J. Phys. Chem. 1994, 98, 11623.
[6] This corresponds to the atoms with the 60, 61 and 65 indexes in the atom order of the input molecule. The description of the constraints applied during geometry optimization is described in the PyRED 'Project.config' configuration file, which is available for download here.
[7] The atoms involved in the rigid-body reorientation algorithm are the following: the atoms with the 63, 1 (i. e. the Fe atom), 60 and 60, 1, 63 indexes for the monomeric heme derivative; with the 1, 5, 4 and 4, 5, 1 indexes for ethane; with the 6, 4, 1 and 1, 4, 6 indexes for propene; with the 5, 8, 11 and 11, 8, 5 indexes for pentanoic acid; with the 10, 7, 4 and 4, 7, 10 indexes for pentanoate and with the 5, 8, 11 and 11, 8, 5 indexes for pentane. The atoms involved in the rigid-body reorientation algorithm are automatically determined by the PyRED program, and their description is available in the PDB files of this R.E.DD.B. project.
[8] C.I. Bayly, P. Cieplak, W.D. Cornell, P.A. Kollman, J. Phys. Chem. 1993, 97, 10269, and http://q4md-forcefieldtools.org/REDServer-Development/resp/.
[9] Thirty-nine atoms are involved in a single intra-molecular charge constraint. The atoms involved in intra-molecular charge constraints are described in the PyRED 'Project.config' configuration file, which is available for download here.
[10] The Mol3 file format: http://q4md-forcefieldtools.org/Tutorial/leap-mol3.php. This file format contains RESP atomic charge values, force field atom types, the notion of head/tail for a molecular fragment, the topology (atom connectivities) of the molecule/molecular fragment as well as the Cartesian coordinates which were optimized by quantum mechanical computations.
[11] J. Wang, P. Cieplak, P.A. Kollman, J. Comp. Chem. 2000, 21, 1049.
[12] F.H. Allen, Acta Cryst. 2002, B58, 380.
[13] J.M. Seminario, Int. J. Quantum Chem. 1996, 60, 1271.
[14] D.A. Case, T.E. Cheatham III, T. Darden, H. Gohlke, R. Luo, K.M. Merz Jr, A. Onufriev, C. Simmerling, B. Wang, R. Woods, J. Comput. Chem. 2005, 26, 1668.
[15] The format for the force field parameter modification file is described at http://ambermd.org/formats.html#frcmod. The corresponding file generated for FPD stereoisomers and analogs can be downloaded here.
[16] PyRED automatically generates a script for the LEaP program (AmberTools). This script has been extended to construct and study numerous FPD-based molecular systems in condensed phase, and is available for download here.
[17] 3*3*3 molecules in the X, Y and Z axes constitute twenty-seven FPD entities.





