
Different charge derivation procedures are handled by the PyRED program:
Each charge derivation procedure is specified with the 'CHR_TYP' keyword (see the System.config configuration file):
Notation: Single point MEP computation//Geometry optimization
-1- RESP-A1
HF/6-31G(d)//HF/6-31G(d) (1) - Connolly surface algo. used in MEP computation - 2 stage RESP fit qwt=0.0005/0.001
Used in the Cornell et al., Kollman et al., Cheatham et al., Wang et al. & Hornak et al. AMBER force fields
-2- RESP-B1
B3LYP/cc-pVTZ SCRF(IEFPCM,Solvent=Ether)//HF/6-31G(d,p) (1), (2) - Connolly surface algo. used in MEP computation - 2 stage RESP fit qwt=0.0005/0.001
Used in the Duan et al. AMBER force field
Be sure to use the Gaussian 2003 program because the IEFPCM solvation model has changed in Gaussian 2009
RESP-B1 is not implemented when using GAMESS or Firefly
-3- RESP-C1
HF/6-31G(d)//HF/6-31G(d) (1) - CHELPG algo. used in MEP computation - 2 stage RESP fit qwt=0.0005/0.001
-4- RESP-O1
HF/6-31G(d)//HF/6-31G(d) (1) - Connolly surface algo. used in MEP computation - 2 stage RESP fit qwt=0.000184
Defined for OPLS force fields (4)
-5- RESP-P1
HF/6-31G(d)//HF/6-31G(d) (1) - CHELPG algo. used in MEP computation - 2 stage RESP fit qwt=0.001184
-6- RESP-A2
HF/6-31G(d)//HF/6-31G(d) (1) - Connolly surface algo. used in MEP computation - 1 stage RESP fit qwt=0.01
-7- RESP-C2
HF/6-31G(d)//HF/6-31G(d) (1) - CHELPG algo. used in MEP computation - 1 stage RESP fit qwt=0.01
Used in the GLYCAM force fields
-8- RESP-X1
b3lyp/6-31G(d)//b3lyp/6-31G(d) (1) - Connolly surface algo. used in MEP computation - 2 stage RESP fit qwt=0.0005/0.001
-9- RESP-Y1
b3lyp/6-31G(d)//b3lyp/6-31G(d) (1) - CHELPG algo. used in MEP computation - 2 stage RESP fit qwt=0.0005/0.001
-10- RESP-X2
b3lyp/6-31G(d)//b3lyp/6-31G(d) (1) - Connolly surface algo. used in MEP computation - 1 stage RESP fit qwt=0.01
-11- RESP-Y2
b3lyp/6-31G(d)//b3lyp/6-31G(d) (1) - CHELPG algo. used in MEP computation - 1 stage RESP fit qwt=0.001
-12- RESP-X11
b3lyp/6-31G(d)//b3lyp/6-31G(d) (1) - Connolly surface algo. used in MEP computation - 3 stage RESP fit qwt=0.0/0.0005/0.001
-13- RESP-Y11
b3lyp/6-31G(d)//b3lyp/6-31G(d) (1) - CHELPG algo. used in MEP computation - 3 stage RESP fit qwt=0.0/0.0005/0.001
-14- RESP-X22
b3lyp/6-31G(d)//b3lyp/6-31G(d) (1) - Connolly surface algo. used in MEP computation - 2 stage RESP fit qwt=0.0/0.01
-15- RESP-Y22
b3lyp/6-31G(d)//b3lyp/6-31G(d) (1) - CHELPG algo. used in MEP computation - 2 stage RESP fit qwt=0.0/0.01
-16- ESP-A1
HF/6-31G(d)//HF/6-31G(d) (1) - Connolly surface algo. used in MEP computation - 1 stage ESP fit qwt=0.0
Used in some AMBER, OPLS & CHARMM force field based simulations
-17- ESP-C1
HF/6-31G(d)//HF/6-31G(d) (1) - CHELPG algo. used in MEP computation - 1 stage ESP fit qwt=0.0
Used in some OPLS & CHARMM force field based simulations
-18- ESP-A2
HF/STO-3G//HF/STO-3G (3) - Connolly surface algo. used in MEP computation - 1 stage ESP fit qwt=0.0
Used in the old Weiner et al. AMBER force field.
-19- ESP-C2
HF/STO-3G//HF/STO-3G (3) - CHELPG algo. used in MEP computation - 1 stage ESP fit qwt=0.0
-20- ESP-X1
b3lyp/6-31G(d)//b3lyp/6-31G(d) (1) - Connolly surface algo. used in MEP computation - 1 stage ESP fit qwt=0.0
-21- ESP-Y1
b3lyp/6-31G(d)//b3lyp/6-31G(d) (1) - CHELPG algo. used in MEP computation - 1 stage ESP fit qwt=0.0
-22- DEBUG
For debugging purposes: do NOT select this keyword. The atomic charge values generated will be rotten!
Information about quantum mechanics (QM) computation implemented in PyRED:
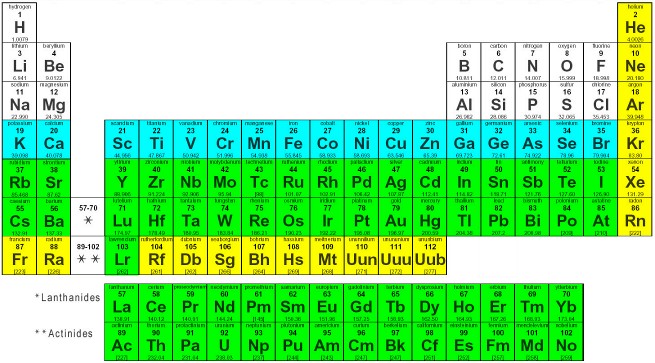
(1) Elements in yellow color are not handled by PyRED. Elements in white (H-Cl; atomic number Z=1 up to Z=17) and blue (K-Br; Z=19 up to Z=35) colors: the 6-31G(d) basis set and the HF method are used by default by PyRED in QM computation as originally defined. Elements in green color (Rb-Lr; Z=37 up to Z=103): the Stuttgart/Dresden and the SBKJC effective core potentials (and associated basis sets) available in the Gaussian and GAMESS/Firefly programs, respectively are used in QM computation. See the 'METHOD_OPTCALC', 'BASSET_OPTCALC', 'METHOD_MEPCALC' and 'BASSET_MEPCALC' keywords in the 'System.config' file to modify the theory level used in QM computation.
(2) Originally developed with the SCRF/IEFPCM solvation model implemented in Gaussian 1998.
(3) Useful only for compatibility with the past (Weiner et al. force field).
(4) Henchman & Essex implemented in the RESP program.
Remark:
van der Waals radii of chemical elements are required in molecular electrostatic potential computation: the radii used to for the Connolly surface were defined by Kollman & Singh, while the radii used in the CHELPG grid of points were defined by Breneman & Wiberg; see also the radii reported by Bondi, they are used by PyRED for transition metals:
- Elements in white color (H-Cl; atomic number Z=1 up to Z=17): element radii originally defined by Kollman & Singh, or Breneman & Wiberg are used by PyRED (see the 'SURFMK_MEPCALC' keyword to affect the Connolly surface if using Gaussian).
- Elements in blue (K-Br; Z=19 up to Z=35) and green (Rb-Lr; Z=37 up to Z=103) colors: element radii were neither defined by Kollman & Singh, nor by Breneman & Wiberg: a generic value of '1.8' is implemented by default in the GAMESS-US/Firefly programs as unknown (see the 'prpel.src', 'prplib.src' and 'svpinp.src' source code files for GAMESS); when the Gaussian program is executed the radii defined by Bondi are used and the '1.8' generic value is used if not available; elements can also be modified by using the 'Element-RAD4MEP' keyword in the Project.config configuration file; see also pieces of general information in Wikipedia.
Last update of this page: June 20th, 2026.
Université de Picardie Jules Verne. Sanford Burnham Prebys Medical Discovery Institute.
© 2009-2026. All rights reserved.

