Introduction
Membrane proteins (MPs) are amphiphilic in nature. As such, to be manipulated and studied in aqueous solution and to remain biologically active, MPs have to be transferred in a suitable membrane-mimicking environment (
i. e. a detergent solution). Unfortunately, there is no systematic way for identifying the ideal detergent for a given MP, as empirical criteria are often used in this undertaking. Among the large variety of detergents available for purifying MPs, n-alkyl phosphocholine detergents, and in particular dodecylphosphocholine (DPC), are considered as good systems to mimic eukaryotic membrane, and thus are widely used to study the structure of transmembrane peptides and MPs.
[1]
Here, we report on the force field topology database (FFTopDB;
i. e. RESP charges embedded in a set of force field libraries in the
Tripos mol2 and
mol3 file formats) for 12 alkyl phosphocholine detergents with linear fatty chains [
i. e. CH2-(CH2)n-CH3, with n = 2-13, see Scheme 1] involved in MP extraction/solubilization experiments. This FFTopDB is compatible with the Cornell
et al. force field and its successive adaptations, and is devoted to condensed phase molecular dynamics simulations.
[2]
Computational details
Charge derivation and force field library building were automatically carried out by using the R.E.D. IV program allowing a rigorous control of the different parameters, which affect charge values compatible with the non-polarizable RESP charge model.
[3] Choline and each alcohol molecular structures were constructed in their extended conformations (
i. e. heavy atom dihedral angles are
trans), whereas ethylmethylphosphate was constructed from the
gauche-gauche conformation of dimethylphosphate available in the "
W-12" R.E.DD.B. project. Geometry optimization and molecular electrostatic potential (MEP) computation were carried out using the
Gaussian 09 program (revision A.02) in the gas phase, while charge fitting was performed using the RESP program.
[4] The Hartree-Fock (HF) method and the 6-31G* basis set were used in geometry optimization, while HF/6-31G* and the Connolly surface algorithm were involved in MEP computation.
The molecular orientation of each optimized geometry was controlled using the rigid-body reorientation algorithm implemented in the R.E.D. program. Two molecular orientations for choline, ethylmethylphosphate and for n-alkyl alcohol molecules (based on the N1C1C2 - C2C1N1, C1C2O1 - O1C2C1 and O1C1C2 - C2C1O1 atom names, respectively) were generated before MEP computation, and involved in the charge fitting procedure to yield reproducible charge values.
[3] Charge fitting was performed using standard two RESP stages with hyperbolic constraint values of 0.0005 and 0.001. The construction of the n-alkyl phosphocholine detergents was carried out from the different molecular fragments obtained by setting required inter-molecular charge constraints during the fitting step involving the hydroxyl group of choline and the methyl group of ethylmethylphosphate, as well as the hydroxyl group of each alkyl alcohol and the ethyl group of ethylmethylphosphate (see Scheme 1). A RRMS (relative root mean square value between MEP values calculated by quantum mechanics, and those generated using the derived charge values) of 0.044 was found for the last fitting stage demonstrating the weak impact of the inter-molecular charge constraints used during charge fitting step.
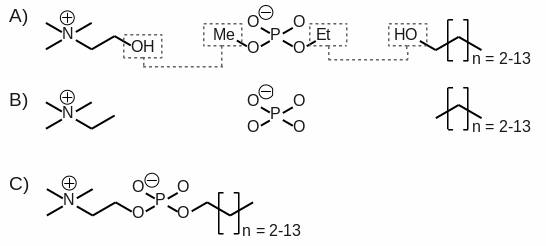 Scheme 1
Scheme 1
A micelle constituted of 54 DPC molecules was constructed. RESP charge values for DPC involved in this micelle were validated in condensed phase over ~150 nsec molecular dynamics simulations using the
Gromacs program (version 4.5.3). Structural features for the DPC micelle obtained from simulations performed using the Hornak force field,
[2] the CHARMM36 as well as different GROMOS force fields were compared.
[5] Good agreements between the different simulations (
i. e. size, shape, DPC headgroup hydration, surfactant conformation, etc...) were observed validating the FFTopDB reported in this work.
References
[1] Arnold & Linke Curr. Protoc. Protein Sci. 2008, 4, Unit 4.8.1–4.8.30; Warschawski et al. BBA Biomembranes 2011, 1808, 1957–1974.
[2] Cornell et al. J. Am. Chem. Soc. 1995, 117, 5179–5197; Hornak et al. Proteins 2006, 65, 712–725.
[3] Dupradeau et al. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839.
[4] Bayly et al. J. Phys. Chem. 1993, 97, 10269–10280, and here.
[5] Klauda et al. J. Phys. Chem. B 2010, 114, 7830–7843; Oostenbrink et al. J. Comput. Chem. 2004, 25, 1656–1676; Poger et al. J. Comput. Chem. 2010, 31, 1117–1125; Schmid et al. Eur. Biophys. J. 2011, 40, 843–856.





