C. Cézard *
Université de Picardie - Jules Verne, Amiens
The purposes of the R.E.D. program and its server R.E.D. Server are to derive RESP or ESP charges and to build force field libraries for small molecules and molecule fragments. Data generated by R.E.D. and R.E.D. Server (charge values, Cartesian coordinates and molecular topology) are available in the Tripos mol2 file format usable as a force field library in any molecular modeling package (AMBER, CHARMM, Gromacs, etc...).
R.E.DD.B. is an online database storing RESP and ESP charge values and force field libraries for small molecules and molecule fragments in the Tripos mol2 file format.
This tutorial describes the use of the
Tripos mol2 file format in the LEaP program available in the AmberTools. To this purpose, new commands have been implemented in LEaP and a perfect compatibility between R.E.D., R.E.DD.B. and LEaP is now observed.
The complete description of these new commands is reported below along with examples.

Using Tripos mol2 force field libraries within LEaP
-I.1- Listing of new commands implemented in LEaP
-I.1.1- Listing of commands available in xLEaP implemented in tLEaP
-I.1.2- Implementation of the savemol2 command in xLEaP and tLEaP
-I.2- Demonstration of the use of these new commands in LEaP in a tutorial

-I.3- Download and install of the LEaP files requested for this tutorial
Using Tripos mol2 force field libraries within LEaP
LEaP actually consists of two programs, the graphical interface xLEaP and its text mode version tLEaP. xLEaP is particularly convenient for building and displaying structures and their related information (atom and residue names, force field atom types, charge values etc...). On the contrary, tLEaP is very efficient for automatically executing scripts in a text mode.
Remark: A new LEaP version (third LEaP version or non-graphical sLEaP) has been added in the AmberTools. However, the features reported in this tutorial are only effective within tLEaP and xLEaP.
-I.1- Listing of new commands implemented in LEaP
The commands available in xLEaP (or tLEaP) can be displayed by typing the "help" command at the prompt message (See the Figure 1 below). All these commands are common to xleap and tleap and thus can be executed in a text mode, i.e. they can be incorporated in scripts.
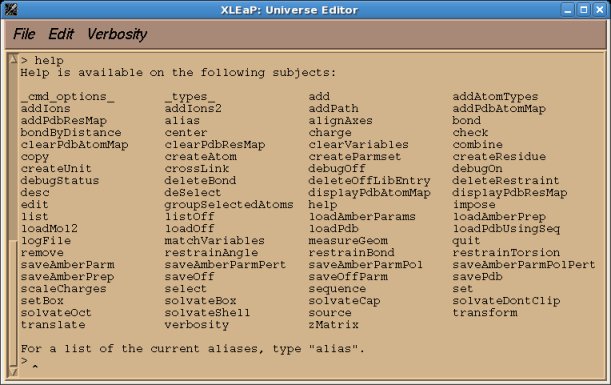
Figure 1
xLEaP features some interesting commands too (See figure 2 below).
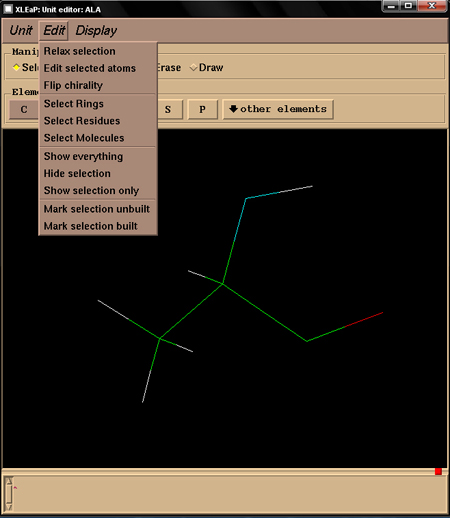
Figure 2
These commands are graphical only and none of them are available in the text mode. Consequently, none of these commands can be used in a script. To solve this problem some of the commands only available in xLEaP have been incorporated in tLEaP.
-I.1.1- Listing of commands available in xLEaP and now implemented in tLEaP
The "relax" command:
This command is used to relax a set of atoms belonging to a unit to remove bad contacts and bumps in a structure. This command is the equal of the "Relax selection" command found in xleap (See Figure 2). Its syntax is the following:
relax unit
The "flip" command:
This command is used to flip the chirality of a center atom. This command is the equal of the "Flip chirality" command found in xleap (See Figure 2). Its syntax is the following:
flip unit
It is important to emphasize that before using one of these new commands, target atoms belonging to the unit have to be selected using the "select" command. Once the relax or flip command has been executed, the selected atoms have to be de-selected with the "deselect" command.
The "select" and "deselect" commands are available in tleap.
-I.1.2- Implementation of the savemol2 command in xLEaP and tLEaP
With the 8th version of the AMBER distribution, an important feature has been added in LEaP: the "loadmol2" command. It is now possible to load a Tripos mol2 file in the LEaP program. However, saving a set of Cartesian coordinates in the Tripos mol2 file format is not possible even in AMBER9. This new command has been implemented in LEaP with the following syntax:
savemol2 unit mol2filename option
The "option" will affect the sixth column of the @<TRIPOS>ATOM group.
"0" will print chemical elements in the column, whereas
"1" will print force field dependant atom types ; if the latters are not available, chemical elements will be printed instead.
This option has been introduced to generate Tripos mol2 file format fully compatible with R.E.DD.B.
-I.2- Demonstration of the use of these new commands in LEaP in a tutorial
The use of the "relax", "flip" and "savemol2" will be illustrated on the central fragment of the Deoxyadenosine nucleotide monophosphate, see Project F-60 for example. The corresponding mol2 file generated by the R.E.D. III.3 program is actually a bare fusion of a Dimethylphosphate and a Deoxyadenosine nucleoside without any relaxation step. The resulting structure is deformed and quite unphysical (see Figure 3a, below). Nevertheless, it has to be noted that in the philosophy of building a force field library such a feature is not a problem since the Cartesian coordinates available in a force field library are not used as a recognition motif. Indeed, a force field library has to be compatible with any type of molecular orientation and/or conformation for a given structure (this statement is also true for enantiomers or diatereoisomers). However, visualizing such a deformed structure is definitely not convenient, and submitting it as is in R.E.DD.B. is not acceptable. The "relax", "flip" and "savemol2" commands can be used in scripts to generate a Tripos mol2 file with a correct set of Cartesian coordinates. The structure corresponding to the Tripos mol2 file obtained after Cartesian coordinate corrections is presented in Figure 3b below.
Remark: This limitation is now solved and the geometry of a fragment originating from the fusion between two molecules is recalculated with R.E.D. III.4 and R.E.D. IV version June 2010.
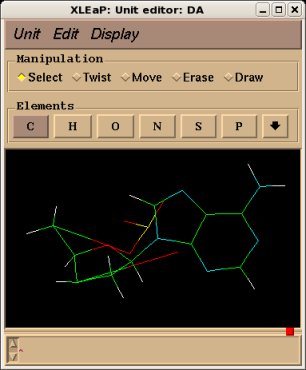 | 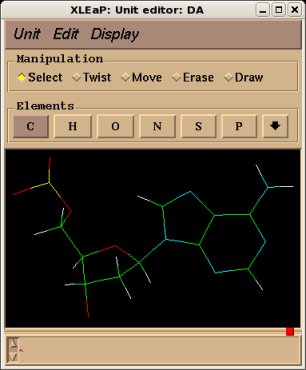 | |
Below is the leap script used to:
(i) load the Tripos mol2 file generated by the R.E.D. III.3 program,
(ii) generate a new set of Cartesian coordinates for the nucleotide fragment, and
(iii) save the new Cartesian coordinates in the Tripos mol2 file format:
# A force field has to be loaded to relax a structure
source leaprc.ff99 # or any other force field
# Load the Tripos mol2 file generated by R.E.D. III.3
DA = loadmol2 ./Frag-DA.mol2
#
# set the FF atom types to obtain a correct structure
set DA.1.P type P
set DA.1.O1P type O2
set DA.1.O2P type O2
set DA.1.O5' type OS
set DA.1.O3' type OS
set DA.1.C5' type CT
set DA.1.H5'1 type H1
set DA.1.H5'2 type H1
set DA.1.O4' type OS
set DA.1.C4' type CT
set DA.1.H4' type H1
set DA.1.C3' type CT
set DA.1.H3' type H1
set DA.1.C2' type CT
set DA.1.H2'1 type HC
set DA.1.H2'2 type HC
set DA.1.C1' type CT
set DA.1.H1' type H2
set DA.1.N6 type N2
set DA.1.H61 type H
set DA.1.H62 type H
set DA.1.C6 type CA
set DA.1.C5 type CB
set DA.1.N7 type NB
set DA.1.C8 type CK
set DA.1.H8 type H5
set DA.1.N9 type "N*"
set DA.1.C4 type CB
set DA.1.N3 type NC
set DA.1.C2 type CQ
set DA.1.H2 type H5
set DA.1.N1 type NC
#
# Select the atoms to be relaxed
select DA.1.O5'
select DA.1.P
select DA.1.O1P
select DA.1.O2P
select DA.1.O3'
#
# relax the atoms selected belonging all to the unit DA
relax DA
#
# deselect the atoms previously selected
deselect DA.1
#
# impose a conformation similar to that found in the AMBER force field topology database
impose DA { 1 } {{ O1P P O5' C5' 60.00 }}
impose DA { 1 } {{ P O5' C5' C4' 180.00 }}
#
# select the center to be flipped
select DA.1.C3'
#
# invert the chirality of the C3' center
flip DA
#
# deselect the atom previously selected
deselect DA.1
#
# select the atoms to be relaxed
select DA.1.O3'
select DA.1.H3'
#
# relax the atoms selected belonging all to the unit DA
relax DA
#
# deselect the atom previously selected
deselect DA.1
#
# Save the new set of cartesian coordinates in the Tripos mol2 file format
# (The force field atom types are not conserved to be compatible with R.E.DD.B.)
savemol2 DA ./Frag-DAbis.mol2 0
LEaP is executed in the text mode i. e. using the tLEaP program (xLEaP can also be used) and the following command:
tleap -f leap-script.cmd This type of LEaP script is now useless with R.E.D. III.4 & R.E.D. IV version June 2010!
-I.3- Download and install of the LEaP files requested for this tutorial
- Before downloading the modified LEaP source code files requested for this tutorial, please check the LEaP license on the AMBER web site. Each user of the LEaP source code files downloaded from q4md-forcefieldtools.org have to strictly follow the license defined on the AMBER website.
- You use the tleap program distributed with the Antechamber package:
(i) Click here to download the modified tleap source code files.
(ii) These modified files will overwrite your original LEaP files. If you want to keep a copy of them, please backup or rename the following files in your $AMBERHOME/src/leap/src/leap directory:
- Makefile
- helptext.text
- commands.c
- pdb.h
- pdb_format.c
- pdb_write.c
- unitio.h
- unitio.c
(iii) Extract the content of the leap-q4md_AC.tgz file in your $ACHOME/tleap directory
(iv) Recompile tleap:
cd $ACHOME/src/leap
make install
- You use the LEaP program distributed with the AMBER8 package:
(i) Click here to download the modified LEaP source code files.
(ii) These modified files will overwrite your original LEaP files. If you want to keep a copy of them, please backup or rename the following files in your $AMBERHOME/src/leap/src/leap directory:
- Makefile
- helptext.text
- commands.c
- pdb.h
- pdb_format.c
- pdb_write.c
- unitio.h
- unitio.c
(iii) Extract the content of the leap-q4md_AMBER8.tgz file in your $AMBERHOME/src/leap/src/leap directory
(iv) Recompile LEaP:
cd $AMBERHOME/src/leap
make install
- You use the LEaP program distributed with the AMBER9 package:
(i) Click here to download the modified LEaP source code files.
(ii) These modified files will overwrite your original LEaP files. If you want to keep a copy of them, please backup or rename the following files in your $AMBERHOME/src/leap/src/leap directory:
- Makefile
- helptext.text
- commands.c
- pdb.h
- pdb_format.c
- pdb_write.c
- unitio.h
- unitio.c
(iii) Extract the content of the leap-q4md_AMBER9.tgz file in your $AMBERHOME/src/leap/src/leap directory
(iv) Recompile LEaP:
cd $AMBERHOME/src/leap
make install
If you have questions about this tutorial, please, send your emails to the q4md-forcefieldtools mailing list. We will answer to the queries about the q4md-forcefield tools in the Amber or CCL mailing lists as well.
Last update of this tutorial page: January 12th, 2012.
Charge derivation data free for download.
Université de Picardie Jules Verne. Sanford Burnham Prebys Medical Discovery Institute.
© 2009-2024. All rights reserved.
